Welcome to the Association for Molecular Pathology's Inherited Disorders webpage. Please note some new accessibility features. Click on terms in blue to open the glossary entry in another tab. Click on any image to enhance the view in another tab. Please contact us with any feedback, questions, or any other requests, please email policy@amp.org.
Happy viewing, The Patient Engagement Subcommittee
An Inherited Disorder is a disease caused in whole or in part by a change in the DNA sequence that is different from the sequence observed in unaffected individuals. This is a difference between an individual's DNA and what we understand to be normal. These differences are called variants or mutations. These disorders can be caused by single mutations, multiple mutations, broad damage to DNA, or a combination of mutations and environmental factors.
During a patient's journey to diagnosis, multiple types of testing may be performed to determine whether they have an inherited disorder and/or which disorder it might be. This webpage has been created by experts in genetics and molecular pathology to provide information and offer additional resources that may help to understand the reports from lab testing the patient may receive during this process. A common type of laboratory testing a patient with an inherited disorder may encounter is DNA sequencing.
DNA sequencing is a procedure used by genetic laboratory professionals that include specialized machines and laboratory techniques to identify the A, C, G, and T bases of the DNA strand. Depending on the specific type of testing being performed (see Diagnostic Testing), either small specific sections of DNA can be sequenced or an entire genome. A specialist physician or doctoral scientist analyzes the data generated through sequencing and prepares a results report that includes the key findings, such as the detection of variants that are associated with an inherited disorder.
What is the difference between getting a gene sequenced and getting the whole genome sequenced?
A gene is a segment of DNA that encodes for a protein in our body that has a particular function. Humans have over 20,000 genes encoded by our DNA, but this only represents 1% of the information in our genome. When diagnostic laboratories use sequencing technology, they can sequence either part of a gene, a single gene, many genes (a “panel”), all or most of the genes in the body (the “exome”), or even the entire genome to make the best diagnosis for their patient. Learn more about the difference between "whole genome sequencing" (WGS) and "whole exome sequencing" (WES) below.
What is gene panel testing?
Gene panel testing is the analysis of multiple genes associated with a inherited disorder or group of disorders. Typically, gene panels are designed to include appropriate genes that can aid in diagnosis. For example, a panel test for an inherited disorder, such as epilepsy might contain over 100 genes that are known to be associated with different inherited disorders that include epilepsy as a symptom. A panel test allows for multiple different genes to be analyzed using a single patient sample.
What is next generation sequencing and why is it different?
Next generation sequencing, or NGS, is a powerful sequencing method that allows a clinical genetics laboratory to sequence an enormous amount of DNA, even an entire genome, at one time. Traditional sequencing (i.e. Sanger sequencing) usually requires multiple reactions to analyze a single gene. NGS technology allows laboratories to run millions of traditional sequencing-like reactions simultaneously. Software stitches the data together and identifies potential mutations. As a result, NGS as a technology has been responsible for the explosion of genetic information over the past decade.
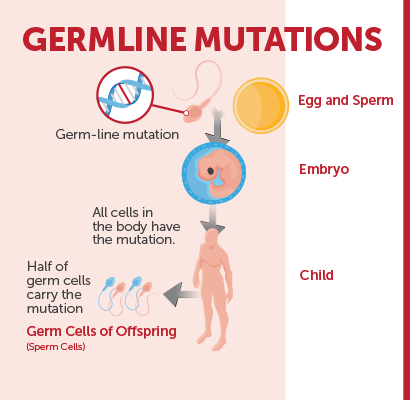
Germline Testing vs Genetic Testing for an Inherited Mutation
AMP would like to acknowledge the phrase “genetic testing for an inherited mutation” is the preferred terminology among patients in reference to germline testing. However, this webpage is built to introduce patients to the information found on their laboratory report and will use those terms to provide the clearest information.
The term ‘germline’ refers to the germ cells (eggs and sperm) which pass on genetic material from generation to generation. Germline testing, frequently referred to as “genetic testing”, detects genetic alterations that were inherited from germ cells and are therefore present throughout a person’s body. Germline testing is typically performed on blood or saliva, not cells or tissue.
A diagnostic germline test may be ordered for a patient with symptoms and/or a strong family history of a genetic disorder to determine the risk to the patient for developing a hereditary disorder. Sometimes a patient may have a diagnosis already, and, a diagnostic germline test is used to determine whether the disorder was inherited. Whether or not an individual has been diagnosed with an inherited disorder, a germline genetic test may provide information about how likely the person might be to pass the disorder on to their children.
Germline testing for inherited disorders is usually considered for three broad categories:
Diagnostic testing may detect or confirm the presence of a genetic disorder.
Carrier screening/testing provides information about the risk for having a child with a genetic disorder and may provide awareness and/or inform decisions about reproduction having children. Carrier screening is often performed for large groups of people (for example, all newborns), even if there is no other reason to be suspicious of the disorder. These tests are highly accurate, but they will sometimes produce an incorrect result. If a screening test is positive, it should always be confirmed with a diagnostic test (see below). If a screening test is negative, there is still some small risk ("residual risk") that the disorder is present.
Newborn screening is universal testing of healthy newborns for rare inherited disorders that can be treated or managed.
More information about diagnostic testing, carrier screening, and newborn screening can be found below in Screening Tests.
Medical professionals administer diagnostic tests to identify a condition in a patient with symptoms or other reason for suspicion, such as close relatives with the condition. Diagnostic genetic testing is used when suspicion for the condition is high. Suspicion might be raised by the patient’s symptoms, their family history, or other laboratory tests. A positive result on a screening test should be confirmed with a diagnostic test. This testing may be done before birth, or after birth to help determine the genetic basis and course of a disorder as well as potential treatment(s).
Diagnostic testing is most often performed for patients with unexplained health problems, to understand the underlying cause, to possibly guide treatment, and support reproductive decisions when applicable. The signs and symptoms a patient experiences may be specific to a certain inherited disorder, in which case testing seeks to confirm the diagnosis. Alternatively, if the signs and symptoms are not specific to a single inherited disorder, testing for multiple disorders with similar features may be performed to determine which specific diagnosis is present. In diagnostic testing, a positive result confirms a specific diagnosis, paving the way for more specific treatment(s).
| Types of Diagnostic Tests | Chromosomal Structural Variants | Methylation issues | Copy number variants (CNV) | Single Nucleotide Variants (SNV) |
|---|---|---|---|---|
| Karyotype |
|
|
|
|
| Fluorescent In Situ Hybridization(FISH) |
|
|
|
|
| Whole Genome Sequencing |
|
|
|
|
| Multiplex Ligation-dependent probe amplification (MLPA) |
|
|
|
|
| Chromosomal Microarray (CMA) |
|
|
|
|
| Polymerase Chain Reaction (PCR) |
|
|
|
|
|
*The techniques that are used for targeted testing are diverse and depend on the size and type of variant(s) being tested |
|
|
|
|
*
The genetic changes that impact inherited disorders vary in size. For example, in some inherited disorders a whole chromosome may be lost or duplicated. Alternatively, a single base pair can be altered and may cause an inherited disorder. Loss of a whole chromosome can be seen with a microscope, but single base changes are too small, and require specific technologies to observe. There is a range of size and complexity of alterations to DNA, and different technologies are used to detect different types of changes.
Imagine you are looking at a newspaper. You would use a different strategy to see if the sports section was missing than you would to look for a typo. Similarly, different types of tests are designed and used to detect changes at different scales. A karyotype uses a light microscope to directly observe chromosome bands to identify “large” genetic changes. These changes are typically millions of base pairs in size. FISH testing uses fluorescent probes with special microscope filters to observe smaller, more specific genetic alterations, typically in the range of hundreds of thousands of base pairs. Microarray technologies (for example: CMA) can vary in identifying changes in DNA from tens of thousands to thousands of base pairs.
Gene sequencing, such as Sanger Sequencing, can identify the smallest changes to DNA, including alterations of a single base pair. They can identify a range of alteration sizes, but the most widely used sequencing technologies have difficulty identifying changes larger than a few hundred bases. Next-Generation Sequencing can be used to examine very large portions of the genome or even the whole genome as well as detect single base changes. Imagine using a microscope to read the newspaper, you would notice all the small typos. However, it would be inefficient to look for a missing sports section which can be identified by eye.
Each technology has additional strengths and weaknesses. FISH, for example, is particularly good at confirming chromosomal rearrangements. Multiplexed Ligation-dependent Probe Amplification (MLPA) is useful for identifying small deletions or duplications.
Diagnostic Tests may be used alone or in different combinations to best diagnose or characterize a particular medical scenario. For example, if a patient has symptoms of trisomy 21 (Down’s Syndrome), karyotyping alone may confirm the diagnosis. On the other hand, if the patient has a nonspecific or unusual combination of symptoms, Next Generation Sequencing of several genes may be performed to identify a genetic cause.
%20(002).png)
A karyotype is a description of how chromosomes appear under a microscope (see image below for an example). Typical human karyotypes are written as either 46, XX or 46 XY. Karyotypes are generated by the examination of microscopic images of chromosomes. These images are arranged as a set called a karyogram (see picture). Karyograms may be produced from any bodily tissue that has living cells, including blood, bone marrow, or fetal cells.
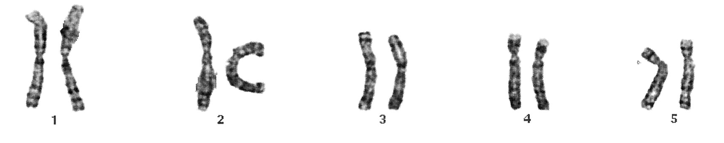
Example above shows only 10 chromosomes. As a note, most indivduals have 46 chromosomes. However, it is not uncommon to have more or less chromosomes
Image from DOI:10.1016/S1525-1578(10)60619-8
To generate a karyotype, the cells are grown in the laboratory. When enough cells are available, the cells are processed on a glass slide. To make the chromosomes more visible, the slides are stained with specific dyes. This causes the chromosomes to show a characteristic banding pattern. The chromosomes are observed and photographed using a microscope, and examined by a professional expert called a cytogeneticist. The cytogeneticist identifies the chromosomes by size and banding pattern, and looks for abnormalities in chromosome number and/or structure. For example, a patient with trisomy 21 (Down’s syndrome) has an extra chromosome 21 in all of their cells, so 47 total chromosomes are observed in their karyogram instead of the usual 46. The Karyotype for a female with trisomy 21 would read as 47, XX +21. Smaller changes to the banding pattern such as loss or gain of a part of a chromosome or a part of a chromosome rearranged to another chromosome can also be observed and described.
How does FISH testing work?
FISH testing uses probes that bind to DNA. The probes are nucleotide sequences that are designed to match a specific target area in human chromosomes, and which also contain fluorescent labels that “glow” (fluoresce) under specific conditions. To perform FISH testing, thin sections of tissue are placed onto glass slides, exposed to the probes, and then analyzed under a fluorescence microscope by trained molecular professionals. These professionals analyze the pattern of fluorescence for any unexpected changes to the chromosomes, and describe what they find. For instance, if parts of chromosomes are deleted, FISH probes labeling the deleted regions will be absent. On the other hand, if parts of the chromosome are duplicated, there will be extra FISH labels for the duplicated regions. FISH can also be used to determine if DNA is in the wrong place (a rearrangement). For all FISH cases, a number ofcells are individually counted and analyzed to determine the overall characteristics of the tissue.
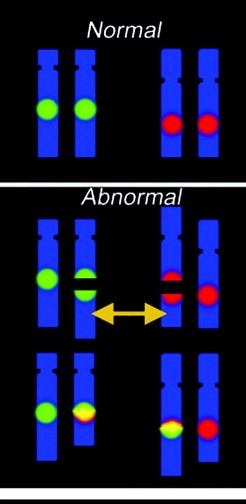
Image from DOI:10.2353/jmoldx.2006.050083
The image shows an example of Fluorescent In Situ Hybridization (FISH) in normal (top) and abnormal (bottom) chromosomes. The probes bind to specific genes or genomic regions (green and red) on different chromosomes. The pattern allows a molecular professional to identify that two chromosomes have switched segments, a translocation event (yellow signal). The new arrangements shown on the bottom may destroy or alter gene function.
What is the difference between FISH testing and having a gene sequenced?
FISH testing uses a microscope to look for the presence, absence, or misplacement of sections of the chromosome. Gene sequencing, on the other hand, determines the “spelling” of an unknown sequence under study and therefore, can detect mutations (“misspellings" of the DNA sequence) as well as pinpointing the exact base-pair position of that DNA change. Sequencing can often detect the alterations that are detected by FISH, but may not be the most efficient option. FISH is often more efficient, but can not detect the spectrum of changes that sequencing identifies. See more about gene alterations in Chromosomal Structural Variants
Whole Genome Sequencing uses Next Generation Sequencing to sequence the majority of a patient’s genome. Some regions are challenging to sequence and may not be included. This approach can be helpful when there are no clear symptoms or family history pointing to a specific inherited disorder.
Whole Exome Sequencing (WES), similar to Whole Genome Sequencing, uses Next Generation Sequencing. However, this test focuses on a patient's exome. Other regions in the human genome that are challenging to sequence or are not associated with known human disease (introns), may not be included in exome sequencing. This approach can be helpful when there are no clear symptoms or family history pointing to a specific inherited disorder.
Click here to access our graphic explaining Whole Genome Sequencing
Multiplex ligation-dependent probe amplification (MLPA) is a multiplex assay to detect copy number variations in DNA sequences. MLPA uses a set of fluorescently labeled primers that bind to specific target sequences in the genome. PCR is performed to make copies of these regions. These PCR products are then separated, measured, and compared to normal control samples. If a sample has a region that has less copies than the controls, we can say that the region is missing or deleted. If a sample has a region with more copies than the controls, then that sample would have a duplication or gain of that specific genomic sequence. With MLPA, more than 40 different DNA sequences can be analyzed in one diagnostic test.
A different version of the test, Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) detects copy number variations (CNVs) like MLPA, and adds the ability to detect changes in methylation, which is important in some medical conditions. If DNA is methylated, this may alter how a gene functions. In the case of MS-MLPA, the patient's DNA is split into two samples. One sample is exposed to a special enzyme that cuts DNA only if it is NOT methylated. The same MLPA test is then performed on both samples. The untreated sample (without the DNA-cutting enzyme) identifies CNVs. Differences between the untreated sample and the treated sample reveal where the DNA was methylated. Thus, MS-MLPA can identify methylation differences as well as copy number differences in a specific DNA region that is being tested.
Chromosomal microarray (CMA) may detect missing or extra copies of regions of DNA. A version of this test called a single nucleotide polymorphism (SNP) microarray can also determine if both the maternal and paternal copies of the region are present or if both copies are from one parent (the copy from the other parent is missing). The test uses DNA obtained from a patient sample which the laboratory breaks into pieces and labels with a fluorescent dye. This DNA is then bound to hundreds of thousands of unique sequences called probes. The DNA will bind only if the probe is an exact match. Specialized equipment is used to measure the amount of signal from the dye when the DNA binds to these probes. The amount of signal will determine if there are missing or extra pieces of DNA in the patient’s sample.
PCR genotyping is a way to quickly detect the presence or absence of variation in DNA. PCR is a chemical reaction that makes thousands of copies of a specific region of DNA enabling detection of changes within the gene being tested. PCR can be modified in many ways to detect a variety of alterations, or even read the exact nucleotides of a patient’s DNA (DNA sequencing). PCR is very direct and efficient for small, specific DNA variations. A doctor may order a PCR test to get results quickly in 1 to 3 days.
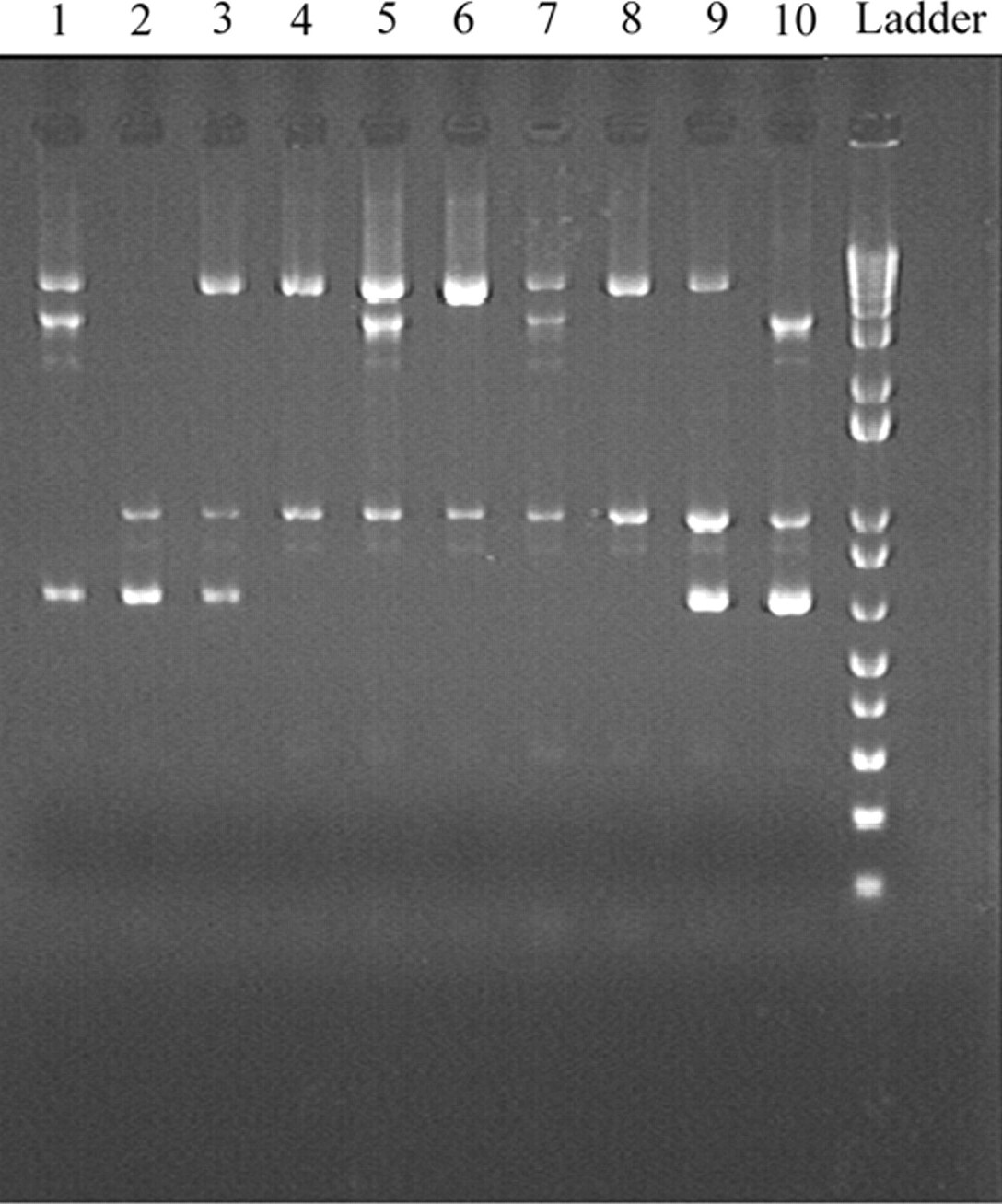
Image from 10.2353/jmoldx.2007.070030
PCR is visualized in a variety of ways through electrophoresis (pictured above)
Another one of the tests used to determine if the gene is correctly marked is methylation-sensitive polymerase chain reaction (MS-PCR) makes millions of copies of DNA that has a specific methylation pattern associated with a disorder. This test can tell us if both parental regions are present or if one is missing.
When a patient has symptoms or family history of a specific inherited disorder, the most efficient testing approach is often to look just at a specific gene, specific regions of a gene, or even a large change in the genome. This can be accomplished using the various methods described above including karyotyping, CMA, and DNA Sequencing. The method of testing is tailored to the size of the DNA alteration and the specific inherited disorder of interest.

Image from "CTSA Resources Support Largest U.S. Newborn Screening Study for Fragile X Mutations" (https://ncats.nih.gov/pubs/features/newborn-fragile-x, Last Accessed January 25, 2023)
Most newborn screening programs are run by the health departments of individual states in the USA. The specific disorders that are tested are not standardized across the country and there is some variation from state to state. However, the purpose of newborn screening is to detect potentially fatal or disabling conditions in newborns as early as possible. This allows for treatment interventions to prevent or reduce the severity of conditions associated with a disorder. Any positive test result from newborn screening should be confirmed by a diagnostic test. Examples of such disorders include congenital hypothyroidism and phenylketonuria. For example, a newborn with a positive screening test result for congenital hypothyroidism should have the result confirmed by a diagnostic test. If the diagnosis is confirmed, prescribing treatment with levothyroxine at an early age can prevent intellectual disability and failure to grow. For a comprehensive list of the newborn screening done across the United State, please click here
Carrier screening is genetic testing performed on people who may display no symptoms of a genetic disorder but may be at risk for passing it on to their children. When a couple is planning a pregnancy, their healthcare provider may recommend carrier screening for “autosomal recessive disorders”. Individuals inherit two copies of the genes located on the autosomes (non-sex chromosomes or chromosomes 1-22) – one from each parent. In many cases, a person only needs one copy of a gene to function properly. Some inherited disorders, such as Sickle Cell Anemia or Cystic Fibrosis, occur when a person inherits two altered copies of a gene (no functional copy). This is known as an “autosomal recessive disorder”. A carrier of an autosomal recessive disorder has one normal and one abnormal version of the gene or allele; often carriers have no signs or symptoms at all. However, if both parents carry alterations for a particular autosomal recessive disorder, then their biological children are at risk for inheriting two abnormal genes (one from each parent) and developing that disorder. An affected individual is a person with a particular trait or disorder.
.png)
Discliamer: AMP would like to acknowledge that the shapes in this pedigree represent biological sex, and not gender identification. We would like to highlight that this is only an example of a pedigree and does not encompass the spectrum of all sex types and gender identities for persons looking for testing.
Prospective parents are the most common candidates for carrier screening and the test usually involves collecting a sample of blood, saliva, or swab from the inside of the cheek. Carrier screening tests can be done before having children or during pregnancy. Often, when when parents are recomended to undergo carrier screening, they will work with a genetic counselor that will create a pedrigree (simliar to the image above) to see relatives who may have been affected, who may have passed on risk for the disorder to their children, and who may be at risk today for having the genetic disorder or having children with the disorder.
Healthcare providers can help parents determine how likely they are to have a child affected with an autosomal recessive disorder. For example, if both parents are carriers of a recessive gene for a disorder, there is a 1-in-4 chance (25%) with each pregnancy that the child will inherit the abnormal gene copy from each parent and will have the disorder. Further, there is an additional 50% chance that a child may be a carrier of the disorder. If only one parent is a carrier for an abnormal copy of a gene, there is a 50% chance that the child will be a carrier of the disorder but a very low risk of being affected with the disorder.
Non-Invasive Prenatal Screening (NIPS, sometimes referred to as NIPT):
For more information regarding Non-invasive Prenatal Screening, a detailed overview of the concepts in terms of laboratory tests, and a comparison of screening versus diagnostic laboratory tests, we invite indivduals to view the recorded session of the NIPS lunch and learn. Alternatively, a written resource on NIPS is available on the Society for Women’s Health Research webpage. This resource walks through the steps a patient will face with NIPS and how to be an advocate.
Genetic laboratory tests are designed to identify differences in a patient's DNA which may be associated with diseases. These differences, sometimes called mutations, but often called variants, are evaluated by laboratory professionals using scientific literature, databases, and software tools. These experts look for associations with inherited (genetic) disorders. When the laboratory determines that a variant contributes to or causes a disorder, the variant is described as “pathogenic”. However, a variant is part of the normal human variation that simply makes people different from each other, it is described as “benign” and will not be mentioned in a laboratory report. A variant is described as a “Variant of Uncertain (or Unknown) Significance” (VUS or VOUS) when it cannot be classified with confidence as either pathogenic or benign.
Importantly, a VUS is just that, a variant of uncertain significance. Any decision, clinical or otherwise – which is based on a VUS finding should be approached with extreme caution. Since a VUS is dependent on existing medical understanding, its classification might change over time as more becomes known about a disorder or a rare genetic variant.
When testing is complete and variants detected have been classified using th e best current knowledge, molecular professionals will send a report describing significant findings to the medical professional who ordered the test for the patient. For more complex testing like whole exome or genome sequencing, providers and genetics professionals will often consult as part of the patient’s care team to connect the genetic test results to what is being seen in the patient. For variants classified as a VUS, testing laboratories will often review the literature and clinical databases periodically to see if more evidence has been reported that can lead to a more helpful classification of the variant. Many laboratories have policies to reevaluate variants previously described as VUS. The policy may be described in the report, and questions should be directed to the laboratory. Ordering providers can also request the performing laboratory to re-review the results. Additionally, the report should provide enough information about the finding to support a second opinion by other health care provider(s). If a change in classification occurs, the laboratory may issue an amendment to the original report that explains the changes.
e best current knowledge, molecular professionals will send a report describing significant findings to the medical professional who ordered the test for the patient. For more complex testing like whole exome or genome sequencing, providers and genetics professionals will often consult as part of the patient’s care team to connect the genetic test results to what is being seen in the patient. For variants classified as a VUS, testing laboratories will often review the literature and clinical databases periodically to see if more evidence has been reported that can lead to a more helpful classification of the variant. Many laboratories have policies to reevaluate variants previously described as VUS. The policy may be described in the report, and questions should be directed to the laboratory. Ordering providers can also request the performing laboratory to re-review the results. Additionally, the report should provide enough information about the finding to support a second opinion by other health care provider(s). If a change in classification occurs, the laboratory may issue an amendment to the original report that explains the changes.
Background
DNA is the hereditary material that encodes all of the information to produce the materials our bodies need to function. DNA is made up of individual bases (A, C, G, and T) and their order, or sequence, determines the instructions encoded by the DNA.
Genes are segments of DNA that instruct the cell how to build proteins. Proteins act as machines and structures that perform many tasks in the body, including "reading" DNA, building other molecules, and interacting with the environment. An exome is every gene together (in humans, there are about 20,000 genes). Genes are arranged in order at specific positions with often long stretches of "non-coding" DNA between them. The DNA between the genes helps control the timing and coordination of genes, but much of its function is still under research. The genome is all of the DNA, including the exome and all the non-coding material. The genome is tightly packed into 23 pairs of chromosomes (46 in total). One chromosome of each pair is inherited from each parent. Of the 23 pairs, 22 are alike in males and females, and are called autosomes. The remaining pair are the sex chromosomes: an X and a Y chromosome in males and two X chromosomes in females. A small amount of DNA is located separately in a special structure called a mitochondria, which is inherited only from the mother.
Single Gene Disorders can also be diagnosed with Targeted DNA Sequencing or Whole Genome Sequencing
DNA Inheritance
DNA is inherited in families through chromosomes. If a gene is altered, the way the altered gene behaves and the type of chromosome where it is positioned cause different inheritance patterns.
Learn more about the inheritance patterns at the links below:
Autosomal Dominant
Autosomal Recessive
Sex- linked
Mitochondrial
X- Linked Dominant
X-Linked Recessive
Molecular diagnostic testing can identify the specific variants in the CFTR gene that cause Cystic Fibrosis. The presence of two abnormal (pathogenic) variants confirms the diagnosis of Cystic Fibrosis, which is an autosomal recessive disorder. Newborn screening for Cystic Fibrosis is in widespread use in the United States, thus the disorder is now often diagnosed before symptoms begin. Specific medicines called CFTR modulators are available as treatment options, depending on which variants are present in the CFTR genes of the patient. Different medications and combinations may only be effective for specific Cystic Fibrosis genetic variants; therefore, the genotyping test results (determining which variants are present) are essential to selecting the best treatments. Carrier screening for specific Cystic Fibrosis genetic variants is also widely performed to identify individuals who are at risk of having children with Cystic Fibrosis. Learn more about Cystic Fibrosis here.
Spinal muscular atrophy is an inherited disorder caused by loss of function of both copies of the SMN1 gene, most often by deletions of part of the gene. Loss of the SMN1 gene’s function leads to loss of SMN protein, which is important for the nerve connections that control muscle function. People have variable numbers of copies of the highly similar gene SMN2, which produces a small amount of the SMN protein. When extra copies of SMN2 are present, enough SMN protein is produced to partially make up for the loss of SMN1 genes, resulting in the less severe forms of spinal muscular atrophy. Testing involves detecting deletions in the SMN1 gene and evaluating the number of copies of SMN2. Spinal muscular atrophy is diagnosed when deletions are present in both copies of the SMN1 gene. The number of SMN2 gene copies helps predict how severe the disorder may be. Other types of testing may be required to detect rare genetic variants that are not due to deletions of SMN1 but still cause the disease. Treatment is tailored to the type of SMA disorder, so accurate testing is very important. Carrier screening is also recommended to identify individuals who are at risk of having children with SMA. Learn more about Spinal Muscular Atrophy here.
Background
Human chromosomes are discrete structures of genetic material within our cells called deoxyribonucleic acid (DNA). Humans have 46 total chromosomes, comprised of 22 autosomal pairs numbered 1-22, and two sex chromosomes, which can be two X chromosomes (biological female) or one X and one Y chromosome (biological male). An individual usually has two copies of each chromosome, one inherited from each parent. Maternal chromosomes come from the mother, while paternal chromosomes come from the father. However it is not uncommon for an indivdual to have more or less chromosomes. Chromosomal abnormalities occur in about 1 in 150 births.
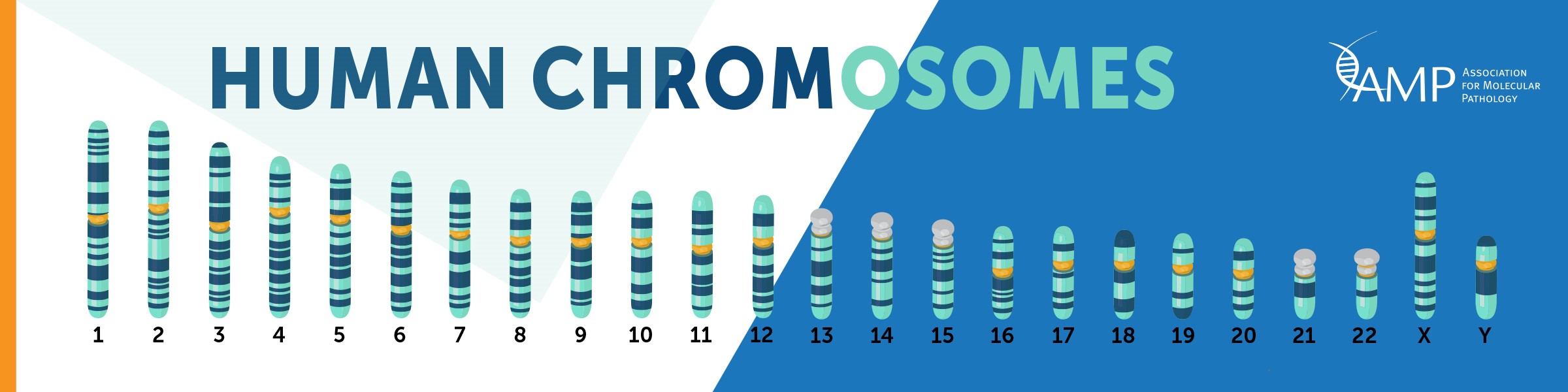
Examples of possible chromosomal abnormalities are below:
| Trisomies | three copies of a chromosome instead of two |
| Monosomies | one copy of a chromosome instead of two |
| Balanced Translocations | parts of chromosomes are swapped but no genetic material is lost |
| Unbalanced Translocations | parts of chromosomes are swapped but genetic material is lost and/or gained |
| Deletions | a chromosome(s) is missing part of its genetic material |
| Duplications | a chromosome(s) contains extra copies of genetic material |
| Inversions | all the expected genetic material is present but in a rearranged order |
| Uniparental Disomy | both copies of a chromosome or part of a chromosome are inherited from the same parent |
Some Diagnostic Tests used to determine if a patient has chromosomal abnormality are Karyotyping, Fluorescent In Situ Hybridization (FISH) or Chromosomal Microarray.
Background on Imprinting:
Generally, both copies of a gene, one inherited from each parent, are active and expressed (i.e. turned “on”). However, for certain genes, only one copy is normally active and expressed, depending on the sex of the parent who contributed the copy. Some genes are only active when inherited from the mother, while other genes may only be active when inherited from the father. This phenomenon is called genomic imprinting, a natural process that uses chemical modifications of DNA (epigenetics) to affect gene inheritance.
Imprinted genes are modified with special chemical “markings” (methyl groups), attached to one copy of the gene during formation of the egg or sperm without changing the DNA sequence. These genes are “marked” differently in females than in males. Methylation markings silence expression of the imprinted gene (turn it “off”). Each person still inherits two copies of the imprinted gene, but only one is expressed (turned “on”). Typically, one copy of the gene is inherited from a parent that is imprinted (methylated or “off”) and one copy is inherited from the other parent that is unmarked (unmethylated or “on”).
Developmental abnormalities and/or disorders may develop when a person inherits either two active copies (“on”), or two inactive copies (“off”) of an imprinted gene. This may randomly occur when a person inherits both copies of an imprinted gene from only one parent (uniparental disomy, UPD) or when there are variants (including deletions) in the genes that affect imprinting.
Examples of disorders associated with imprinting errors are Prader-Willi and Angelman syndromes and certain cancers such as Wilms tumor.
How are Imprinted Disorders Diagnosed?
Diagnosis can be made through a blood test or through a tissue biopsy that is then sent for molecular diagnostic testing. Molecular diagnostic tests for suspected imprinting related disorders can include:
List of Relevant tests
Some imprinting disorders are caused by variants in genes that are imprinted. For example, variants in the UBE3A gene cause Angelman syndrome only if the variant was inherited from the mother. If the variant was on the copy inherited from the father, the individual would not have the disorder. This is because the maternal UBE3A gene is the only functional copy in the brain (the paternal copy is turned off in the brain). To detect these variants, DNA sequencing of the gene is performed.
Another example is sequencing of the CDKN1C gene when the clinician suspects Beckwith-Wiedemann syndrome. Parental testing may be performed to confirm whether the parent carries the variant or not. The patient may have obtained the variant de novo, which means that the variant is new in the patient and the parents do not have the variant in their non-germline cells.
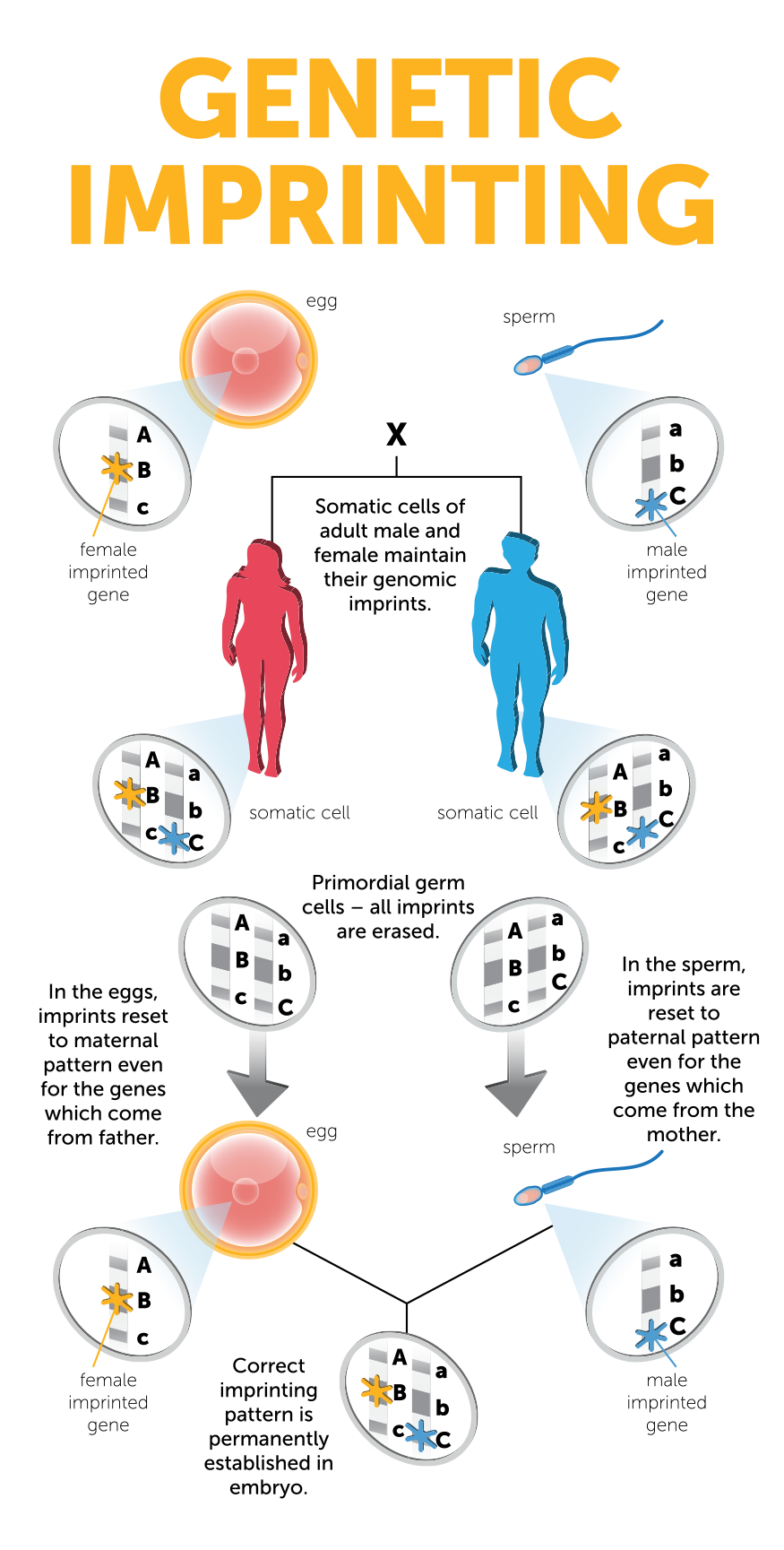
The image above shows how the genetic imprinting pattern is erased and then reset throughout generations. Virtually every somatic cell in an individual’s body has a specific imprinting pattern throughout the genome, with one copy of their genes imprinted in a pattern inherited from their biological father and the other copy of their genes imprinted in a pattern inherited from their biological mother. As sex cells, eggs and sperm, are developed the precursor cells – known as primordial germ cells – have their genetic imprinting pattern erased. In a female, the primordial germ cell goes on to become an egg and the genetic imprinting pattern resets across both sets of genes to match the maternal genetic imprinting pattern (the general female imprinting pattern). In a male, the primordial germ cell goes on to become a sperm cell and the genetic imprinting pattern resets across both sets of genes to match the paternal genetic imprinting pattern (the general male imprinting pattern show in the image). This process ensures that when an egg and sperm cell fuse, the resulting offspring has a balance of both the maternal and paternal imprint pattern.

Most of the genetic testing for diagnosis of inherited disorders is performed on DNA from the nucleus of the cell (the chromosomes). However, there are some rare disorders caused by variants in mitochondrial DNA (mtDNA). Mitochondria, which exist in the cell, but outside the nucleus, have their own small DNA genome. We inherit our nuclear, chromosomal DNA from both of our parents (one set of chromosomes from our father and one set from our mother). In contrast, all of our mtDNA is inherited from our mother. There are many mitochondria in each of our cells, therefore 1000s of copies of mtDNA may exist in each cell. In many cases, only some of the mtDNA in a cell will have variants, a concept called heteroplasmy. There are many different genetic mitochondrial disorders that can occur when there are variants in the mitochondrial DNA (mtDNA). In some cases, mitochondrial disorders can result from a new (de novo) mtDNA variant in a person with no family history of the disorder.
Molecular laboratories employ special techniques to isolate mtDNA from a blood or tissue sample to test for mtNA variants, either by DNA sequencing or by other PCR-based variation detection techniques.
Some inherited disorders are caused by genetic variations of the sex chromosomes. Sex chromosomes come in two different forms, X and Y, unlike the 22 pairs of autosomes. A biological female individual has two copies of the X chromosome (one inherited from each parent), and a biological male individual has one copy of an X chromosome (inherited from his mother) and one copy of a Y chromosome (inherited from his father). In female cells, one copy of the X chromosome is silenced by X chromosome inactivation. This makes sure that male and female cells operate from the same number of X chromosomes. The Y chromosome is quite small (see picture) and does not contain many genes.
An inherited disorder which is “sex-linked” affects a gene on one of the sex chromosomes.Males are more likely to be affected by X-linked disorders because they have only one copy of an X-linked gene. There is no unaffected (normal) gene copy to balance a mutation, which that male inherited from his mother. If a female inherits a mutated copy of an X-linked gene, the unaffected (normal) copy may be active in many of her cells. Thus, a female is more likely to be a carrier of a sex-linked disorder and less likely to be affected. A female carrier of an X-linked disorder has one copy of the abnormal gene and can pass it on to her children, but she usually has milder symptoms of the disorder, or none at all. Male children are affected with an X-linked disorder when they inherit an abnormal X chromosome from their mother that contains the mutated gene or region. Women with a family history of sex-linked disorders may seek prenatal testing to determine the risk of their fetus inheriting the disorder.
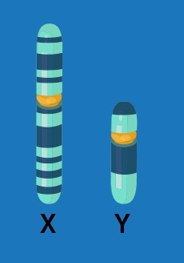
How are Sex Linked Disorders Diagnosed?
Duchenne Muscular Dystrophy (DMD) is caused by changes in the DMD gene. This gene produces the protein dystrophin, which maintains the membrane of muscle cells. If there is a family history of DMD, a single gene test will be used to examine the DMD gene. However, when there is no family history or the genetic variant is unknown, then a broader test (e.g. CMA or MLPA) will be used to detect large deletions or duplications of the DMD gene. About one-third of patients with DMD have no prior family history, occurring for the first time in that patient (“index case”) with a unique variant that was not inherited from either parent (“de novo” variants). Treatment of DMD requires the use of braces, walkers and wheelchairs to maintain mobility. Screening for cardiac or heart issues is necessary because certain heart medications like beta blockers can help prevent heart muscle deterioration. Some variants in the DMD gene may also cause heart issues in carrier mothers so screening may be recommended in these individuals as well. Therefore, early testing is crucial to prevent muscle weakening. Learn more about Duchenne Muscular Dystrophy here.
Fragile X syndrome (FXS) is caused by an increased number of triplet repeats (simple sequences of DNA that are repeated many times) in the gene Fragile X Messenger Ribonucleoprotein 1 (FMR1) on the X chromosome. The large number (over 200) of these repeats causes the FMR1 gene to be shut off which leads to loss of the protein, FMRP. FMRP is essential in the brain, testes, and ovaries, and its loss leads to a specific set of symptoms (a “syndrome”). A specific PCR test can identify the length of the repeated section in the FMR1 gene and provide a diagnosis of FXS. This test can also detect individuals who are at risk for having a child with FXS. Fragile X Syndrome affects both males and females. However, females usually have less severe symptoms because they inherit two copies of the X chromosome and X inactivation reduces the effect of an altered copy. Treatment for FXS consists of therapy and medication to treat the symptoms of the disorder. Testing early is essential in achieving early intervention, as that will give a person with FXS the best start in life. Learn more about Fragile X Syndrome here.
AMP has many Molecular in My Pocket cards related to a number of inherited disorders, including: Hearing Loss, Cystic Fibrosis, Duchenne Muscular Dystrophy/Becker Muscular Dystrophy, Prothrombin-related Thrombophilia, Factor V Leiden, Sickle Cell Anemia, Trinucleotide Repeat Disorders, Prader-Willi and Angelman.
Access it here:
Created: 3/2023